
Over the last several years, there’s been a demand for higher potency cannabis products. Recently, manufacturers have developed the methodology to purify certain cannabinoids, typically THC-A and CBD-A, into their ultra-pure crystalline form. These products are then sold directly to consumers and marketed as “Diamonds” and other unique marketing naming conventions. Prior to going to market, these products are tested and can sometimes result in labs results over 100% purity.
This has led to the question:
“How can my product contain more that 100% THC-A?”
Before we answer that, we would like to present some background information that will help explain how this can be possible.
Brief History
Historically, chemical compounds with 100% purity were only accessed by professionals in scientific communities. Products manufactured to 100% purity and offered as consumer goods and products has been very limited prior to the inception of crystalline forms of cannabinoids. Not even 190 proof grain alcohol on the shelves of your local liquor store is 100% ethanol. Rather, products are manufactured to a target value with a percentage of uncertainty around that target.
While still in its infancy, high-purity cannabinoid products might eventually catch the attention of the FDA. On Friday April 13, 2018, the FDA announced steps to regulate “highly concentrated and pure” caffeine products.
Today, the U.S. Food and Drug Administration took an important step to better protect consumers from the dangers of highly concentrated and pure caffeine products. These products present a significant public health threat because of the high risk that they will be erroneously used at excessive, potentially dangerous doses. Highly concentrated and pure caffeine, often sold in bulk packages, have been linked to at least two deaths in otherwise healthy individuals.
https://www.fda.gov/NewsEvents/Newsroom/PressAnnouncements/ucm604485.htm
Pharmaceutical Landscape
The Pharmaceutical industry, one of the most tightly controlled industries in the world, manufactures products to a target value of plus or minus (±) a certain percent based on criteria for that product type. The nomenclature of a pure drug substance is also known as Active Pharmaceutical Ingredient (API)1; the Potency is referred to as an Assay value. Assay values that exceed 100% are not uncommon in the Pharmaceutical industry where formulators use pure API drug substances to which they add inactive ingredients, referred to as an excipient2, to arrive at a target concentration of a formulated drug product. If a formulator uses an API with an Assay value of, say 101.2% for example, that formulator uses 101.2% in his/her calculation to formulate the product to a given target. This is such a normal and ubiquitous occurrence that the FDA expects to see Assay values for API’s and other chemical standards that exceed 100%.
United States Pharmacopiea (USP)
In the Pharmaceutical industry, many aspects of testing are outlined by the United States Pharmacopeia (USP), a non-profit entity that publishes standards for ingredient, products, and processes. They publish current versions of these standards annually.
The USP–NF is a combination of two compendia, the United States Pharmacopeia (USP) and the National Formulary (NF). It contains standards for medicines, dosage forms, drug substances, excipients, biologics, compounded preparations, medical devices, dietary supplements, and other therapeutics. The current version of USP–NF standards deemed official by USP are enforceable by the U.S. Food and Drug Administration for medicines manufactured and marketed in the United States. (http://www.uspnf.com/purchase-usp-nf)
The USP lists standards specific to purified substances (e.g. ascorbic acid, ibuprofen, pseudoephedrine, etc.) as well as the drug product formulated from these substances are in what are called monographs. While the USP is a non-governmental organization, the FDA considered these monographs (and other items contained in the current USP) as enforceable rule. The USP monographs describe the minimum tests substances or product must meet to be compliant in the United States. An example of a USP monograph for Ibuprofen is presented below.
USP Monograph for Ibuprofen - Read more
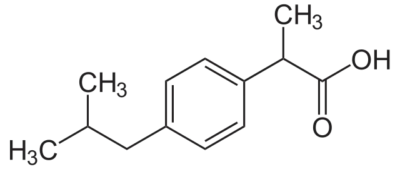
(±) Mixture [58560-75-1].
» Ibuprofen contains not less than 97.0 percent and not more than 103.0 percent of C13H18O2, calculated on the anhydrous basis. Packaging and storage— Preserve in tight containers. USP Reference standards — USP Ibuprofen RS. Identification— A: Infrared Absorption — Do not dry specimens. B: Ultraviolet Absorption —Solution: 250 µg per mL. Medium: 0.1 N sodium hydroxide.
Respective absorptivities at 264 nm and 273 nm, calculated on the anhydrous basis, do not differ by more than 3.0%.
C: The chromatogram of the Assay preparation obtained as directed in the Assay exhibits a major peak for ibuprofen, the retention time of which, relative to that of the internal standard, corresponds to that exhibited in the chromatogram of the Standard preparation, obtained as directed in the Assay.
Water, Method I : not more than 1.0%. Residue on ignition : not more than 0.5%. Heavy metals, Method II : 0.002%. Chromatographic purity—
Mobile phase— Prepare a suitable filtered mixture of water, previously adjusted with phosphoric acid to a pH of 2.5 and acetonitrile (1340:680). Make adjustments if necessary (see System Suitability under Chromatography ). Test preparation— Prepare a solution of Ibuprofen in acetonitrile containing about 5 mg per mL.
Resolution solution— Prepare a solution in acetonitrile containing in each mL about 5 mg of Ibuprofen and 5 mg of valerophenone.
Chromatographic system (see Chromatography)— The liquid chromatograph is equipped with a 214-nm detector and a 4-mm × 15-cm column that contains 5-µm packing L1 and is maintained at 30 ± 0.2 . The flow rate is about 2 mL per minute. Chromatograph a series of 5-µL injections of the Test preparation to condition the column. Chromatograph the Resolution solution, and record the peak responses as directed for Procedure: the relative retention times are about 0.8 for valerophenone and 1.0 for ibuprofen, and the resolution, R, between the valerophenone peak and the ibuprofen peak is not less than 2.0.
Procedure— [NOTE—Use peak areas where peak responses are indicated.] Inject about 5 µL of the Test preparation into the chromatograph, record the chromatogram, and measure the peak responses. Calculate the percentage of each impurity taken by the formula: 100ri / rt, in which ri is the response of an individual peak, other than the solvent peak and the main ibuprofen peak, and rt is the sum of the responses of all the peaks, excluding that of the solvent peak: not more than 0.3% of any individual impurity is found, and the sum of all the individual impurities found does not exceed 1.0%.
Organic volatile impurities, Method V : meets the requirements. Solvent— Use dimethyl sulfoxide.
Limit of 4-isobutylacetophenone— Using the chromatograms of the Assay preparation and the 4-Isobutylacetophenone standard solution obtained as directed in the Assay, calculate the percentage of 4-isobutylacetophenone (C12H16O) in the portion of Ibuprofen taken by the formula: 10,000(C / W)(RU / RS), in which C is the concentration, in mg per mL, of 4-isobutylacetophenone in the 4- Isobutylacetophenone standard solution; W is the weight, in mg, of Ibuprofen taken to prepare the Assay preparation; and RU and RS are the peak response ratios of 4- isobutylacetophenone to valerophenone obtained from the Assay preparation and the 4- Isobutylacetophenone standard solution, respectively: not more than 0.1% is found. Residual solvents : meets the requirements. (Official January 1, 2007)
Assay—
Mobile phase— Dissolve 4.0 g of chloroacetic acid in 400 mL of water, and adjust with ammonium hydroxide to a pH of 3.0. Add 600 mL of acetonitrile, filter, and degas. Make adjustments if necessary (see System Suitability under Chromatography ). Internal standard solution— Prepare a solution of valerophenone in Mobile phase having a concentration of about 0.35 mg per mL. Standard preparation— Dissolve an accurately weighed quantity of USP Ibuprofen RS in Internal standard solution to obtain a solution having a known concentration of about 12 mg per mL.
4-Isobutylacetophenone standard solution— Quantitatively dissolve an accurately weighed quantity of 4-isobutylacetophenone in acetonitrile to obtain a solution having a known concentration of about 0.6 mg per mL. Add 2.0 mL of this stock solution to 100.0 mL of Internal standard solution, and mix to obtain a solution having a known concentration of about 0.012 mg of 4-isobutylacetophenone per mL.
Assay preparation— Transfer about 1200 mg of Ibuprofen, accurately weighed, to a 100-mL volumetric flask, dilute to volume with Internal standard solution, and mix.
Chromatographic system (see Chromatography)— The liquid chromatograph is equipped with a 254-nm detector and a 4.6-mm × 25-cm column that contains packing L1. The flow rate is about 2 mL per minute. Chromatograph the Standard preparation, and record the peak responses as directed for Procedure: the resolution, R, between the ibuprofen and internal standard peaks is not less than 2.5, and the relative standard deviation for replicate injections is not more than 2.0%. Chromatograph the 4-Isobutylacetophenone standard solution, and record the peak responses as directed for Procedure: the relative retention times are about 1.0 for valerophenone and 1.2 for 4-isobutylacetophenone, the tailing factors for the individual peaks are not more than 2.5, the resolution, R, between the valerophenone peak and the 4-isobutylacetophenone peak is not less than 2.5, and the relative standard deviation for replicate injections is not more than 2.0%.
Procedure— Separately inject equal volumes (about 5 µL) of the Standard preparation, the Assay preparation, and the 4-Isobutylacetophenone standard solution into the chromatograph, record the chromatograms, and measure the responses for the major peaks. The relative retention times are about 1.4 for the internal standard and 1.0 for ibuprofen. Calculate the quantity, in mg, of C13H18O2 in the portion of Ibuprofen taken by the formula: 100C(RU / RS), in which C is the concentration, in mg per mL, of USP Ibuprofen RS in the Standard preparation; and RU and RS are the peak response ratios obtained from the Assay preparation and the Standard preparation, respectively. Auxiliary Information— Staff Liaison : Clydewyn M. Anthony, Ph.D., Scientist
One thing that should be immediately apparent is that the Assay specification for this substance, Ibuprofen, is between 97.0% and 103.0%.
Additional examples of other USP monographs with specifications above 100%:
- Pseudoephedrine Hydrochloride contains not less than 98.0 percent and not more than 100.5 percent of C10H15NO·HCl, calculated on the dried basis.
- Anhydrous Citric Acid contains not less than 99.5 percent and not more than 100.5 percent of C6H8O7, calculated on the anhydrous basis.
- Clarithromycin contains not less than 96.0 percent and not more than 102.0 percent of C38H69NO13, calculated on the anhydrous basis.
Question Answered
How can my product contain more that 100% THC-A and how can the Pharmaceutical products described above contain more than 100%?
Answer: The accumulation of uncertainty.
Theoretically, you can’t have more than 100% of anything. Scientifically, when you combine the uncertainty of the 1) Chemical Standards, 2) Laboratory Processes, 3) Laboratory Equipment, and 4) Laboratory Personnel, the additive effect of uncertainty translates into the final Assay value which can derive values in excess of 100% for ultra-pure compounds.
The goal of any laboratory is to minimize the uncertainty to the best of their scientific ability.
Alleviation of Confusion
Values greater than 100% in the scientific community are simply accepted. Chemists, formulators, management, FDA auditors, and the like all understand this concept and have all accepted it for decades. A new sector of talent is entering this scientific community, the Cannabinoid Purification Manufacturer, who in most cases have not been introduced to this concept. To make matters more challenging, manufacturers are marketing these products directly to consumers. Marketing products that contain more than 100% THC-A can create confusion for your end-user.
Remember from our discussion above, never before has a consumer product been manufactured, purified, and sold on the open market directly to consumers. And there’s the rub. If this were being sold to scientific professionals who intend to manufacture products from this as a starting material, they would simply use that Assay value in their formulations.
For this reason, Agricor Laboratories suggests seeking approval with your internal compliance team, your legal counsel, and the MED to label your products that result in Potency values in excess of 100% to an even 100%.
About Agricor Laboratories
Agricor Laboratories is a State-certified marijuana testing laboratory based in Denver, Colorado. Agricor’s mission is to provide the most accurate, transparent, and consistent laboratory services for Colorado marijuana cultivators and infused product manufacturers.
Agricor Laboratories will give you the peace of mind that you’re getting the right analytical answer. Our methods and processes have been thoroughly validated to ensure accuracy and repeatability in the data we provide. We’re ISO 17025 certified and follow current Good Manufacturing Practices and Good Laboratory Practices.
To learn more, feel free to give us a call at (720) 460-3489 or contact us at support@agricorlabs.com.
- Any substance or mixture of substances intended to be used in the manufacture of a drug (medicinal) product and that, when used in the production of a drug, becomes an active ingredient of the drug product. Such substances are intended to furnish pharmacological activity or other direct effect in the diagnosis, cure, mitigation, treatment, or prevention of disease or to affect the structure or function of the body.
- An excipient is a substance formulated alongside the active ingredient of a medication, included for the purpose of long-term stabilization, bulking up solid formulations that contain potent active ingredients in small amounts, or to confer a therapeutic enhancement on the active ingredient in the final dosage form, such as facilitating drug absorption, reducing viscosity, or enhancing solubility.
- In the context of Food and Drug Administration (FDA) regulation, monographs represent published standards by which the use of one or more substances is automatically authorized.